กะโหลกศีรษะเชื่อมติดผิดปกติ
Craniosynostosis
ศ. นพ.นนท์ โรจน์วชิรนนท์
4 ม.ค. 68
ภาวะนี้เกิดจากการที่กระดูกที่มาประกอบกันเป็นกะโหลกศีรษะมีการเชื่อมติดกันผิดปกติตรงตำแหน่งที่เป็นรอยประสาน (cranial suture) ทำให้กระดูกทั้งสองข้างของรอยประสานไม่สามารถขยายขนาดรองรับสมองที่เติบโตใหญ่ึ้น ตามมากด้วยกดรัดสมองที่อยู่ด้านล่าง
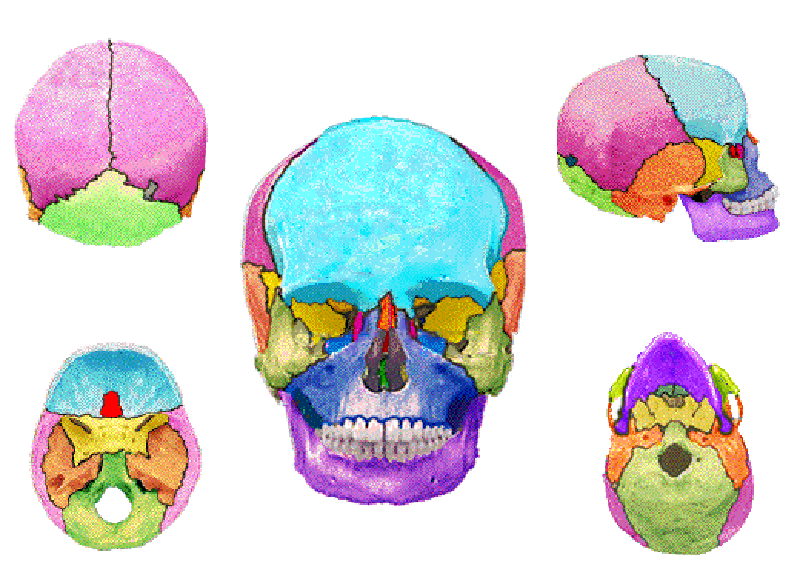
ความผิดปกติดังว่านี้ เกิดขึ้นตั้งแต่ตอนที่ทารกอยู่ในครรภ์มารดา และเกิดต่อเนื่องภายหลังจากคลอดออกมาแล้ว แต่มักจะไม่สามารถตรวจเห็นได้ด้วยอัลตราซาวน์ที่นิยมทำกันในช่วงตั้งครรภ์ แม้แต่เมื่อเกิดมาแล้ว แพทย์ก็อาจวินิจฉัยไม่ได้ถ้าไม่มีความเชี่ยวชาญหรือคุ้นเคยกับโรคในกลุ่มนี้
ประเภทของโรค
โรคในกลุ่มนี้แบ่งออกได้เป็น 2 ประเภท คือ
1. กลุ่มที่มีการเชื่อมติดของรอยประสานกะโหลกศีรษะหลายตำแหน่ง (multiple-sutured craniosynostosis)
มักพบร่วมกับความผิดปกติอวัยวะอื่น ๆ ในร่างกาย เรียกว่า syndromic craniosynostosis เช่น กลุ่มอาการครูซอง (Crouzon Syndrome, รูปที่ 2), กลุ่มอาการเอเพิร์ต (Apert Syndrome, รูปที่ 3), กลุ่มอาการไฟเฟอร์ (Pfeiffer Syndrome, รูปที่ 4), กลุ่มอาการมูนคู (Muenke syndrome), กลุ่มอาการเซเตอร์-โชเซน (Satre-Chotzen Syndrome), กลุ่มอาการคาร์เพนเตอร์ Carpenter Syndrome)


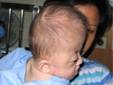




ผู้ป่วยเหล่านี้มีการเชื่อมติดของกระดูกกะโหลกศีรษะหลาย ๆ ชิ้น ทำให้มีกะโหลกศีรษะและใบหน้าผิดรูปอย่างรุนแรงไปพร้อม ๆ กัน มักมีต้นเหตุจากการหลายพันธุ์ของยีน ทำให้มีความพิการแต่กำเนิดของอวัยวะอื่น ๆ ของร่างกายร่วมด้วย เช่น แขน ขา มือ เท้า หัวใจ กระดูกสันหลัง อวัยวะการได้ยิน กล้ามเนื้อตา ระบบต่อมไร้ท่อ
ด้วยเหตุที่ ผู้ป่วยส่วนใหญ่ได้รับยีนที่มีการกลายพันธุ์จากพ่อหรือจากแม่ในช่วงของการสร้างอสุจิหรือไข่ หรือระหว่างปฏิสนธิ หรือระหว่างตั้งครรภ์ ทำให้ผู้ป่วยส่วนใหญ่เป็นโรคขึ้นมาเป็นคนแรกในครอบครัว
และด้วยความรู้เรื่องยีนต้นเหตุที่มีมากขึ้น พบว่ามีโรคหลาย ๆ โรคเกิดจากการกลายพันธุ์ของยีนเดียวกัน จึงเริ่มมีการเรียกชื่อโรคตามยีนที่มีการกลายพันธุ์มากขึ้น เช่น FRFR2-related craniosynostosis หลาย ๆ โรคที่เคยวินิจฉัยไว้ ก็พบว่าผิดพลาดกลายเป็นอีกโรคหนึ่ง
ในส่วนของใบหน้าและศีรษะ การเชื่อมติดผิดปกติของกระดูกหลาย ๆ ตำแหน่ง มีผลที่สำคัญ 3 ประการ คือ
- กะโหลกศีรษะตีบรัดสมอง (craniostenosis) - เป็นผลจากการที่กะโหลกศีรษะไม่สามารถขยายตัวได้ตามปกติ เกิดการกดรัดสมองซึ่งเจริญเติบโตอย่างรวดเร็วในช่วงอายุ 2-3 ขวบแรก ผู้ป่วยอาจมีปัญหาทางด้านพัฒนาการและสติปัญญาได้ ศีรษะมักมีรูปลักษณะผิดปกติ เช่น เป็นรูปทรงสูง (turricephaly) รูปทรงแหลมขึ้นไปตรงกลางศีรษะ (oxycephaly) แบนในแนวหน้าหลัง (brachycephaly) หรือบางครั้งรุนแรงจนศีรษะบิดเบี้ยวอย่างมากรูปร่างคล้ายดอกจิก (cloverleaf skull)
- กระดูกเบ้าตาเล็กผิดปกติ (orbitostenosis) - กระบอกตาประกอบด้วยกระดูกหลายชิ้นประสานกันอยู่ หากมีการเชื่อมติดผิดปกติ ก็จะไม่สามารถขยายขนาดตามลูกตาที่โตขึ้น เป็นผลให้ลูกตาซึ่งมีขนาดใหญ่ขึ้นตามปกติไม่มีที่อยู่ จึงมองเห็นโปนโต (exorbitism) ในรายที่เป็นรุนแรง จะหลับตาไม่สนิทหรือถลนหลุดออกนอกเบ้า จนเกิดเป็นแผลที่กระจกตา(ตาดำ)และตาบอดได้ในที่สุด
- ใบหน้าส่วนกลางเล็กผิดตปกติ (faciostenosis) - กระดูกใบหน้าก็ประกอบด้วยกระดูกหลายชิ้นประสานกัน เมื่อมีการเชื่อมติดผิดปกติ โดยเฉพาะอย่างยิ่งส่วนที่ติดกับฐานกะโหลกศีรษะ ใบหน้าส่วนที่ติดกันคือใบหน้าส่วนกลางจึงไม่ขยายขนาดตามปกติ มีลักษณะเว้าแบน ฟันบนอยู่หลังต่อฟันล่าง มีเพดานปากที่สูงกว่าปกติ (high-arched palate) โพรงจมูกเล็กจน หายใจลำบากจนขาดอ็อกซิเจนได้
| ชื่อโรค | รอยประสานที่มักผิดปกติ | อาการแสดง |
|---|---|---|
| ถ่ายทอดทางกรรมพันธุ์แบบยีนเด่น (autosomal dominant) | ||
| กลุ่มอาการครูซอง (Crouzon) | โคโรนอล (coronal), แซกจิตัล (sagittal) | กะโหลกศีรษะรูปร่างผิดปกติ ใบหน้าส่วนกลางไม่เจริญ ตาโปน กระบอกตาห่างกัน โพรงจมูกตีบแคบ |
| กลุ่มอาการเอเพิร์ต (Apert) | coronal, sagittal, แลมดอย (lambdoid), etc | กะโหลกศีรษะรูปร่างผิดปกติ ใบหน้าส่วนกลางไม่เจริญ ตาโปน กระบอกตาห่างกัน ทางเดินหายใจแคบ นิ้วมือนิ้วเท้าติดกัน |
| กลุ่มอาการไฟเฟอร์(Pfeiffer) | ||
| ชนิดที่ 1 | coronal, sagittal | กะโหลกศีรษะรูปร่างผิดปกติคล้ายกลุ่มอาการเอเฟิต แต่มีหัวแม่มือและแม่เท้าใหญ่ผิดปกติ นิ้วมือและเท้าอาจติดกันได้บ้างแต่ติดเฉพาะผิวหนัง |
| ชนิดที่ 2 | ทุกรอยต่อ | เหมือนชนิดที่ 1 แต่โรครุนแรงกว่า ศีรษะเป็นรูปดอกจิก (cloverleaf หรือ Kleeblattschadel) ปัญญาอ่อน ข้อศอกติด |
| ชนิดที่ 3 | ทุกรอยต่อและรุนแรงตั้งแต่เกิด | ตาโปนมาก กระบอกตาเล็กตื้น ใบหน้าส่วนกลางเว้าอย่างมาก ข้อศอกติดงอเหยียดไม่ได้ อายุสั้น |
| กลุ่มอาการเซเตอร์-โชเซ็น (Saethre-Chotzen) | coronal | กะโหลกศีรษะรูปร่างผิดปกติ มักจะแบนในแนวหน้าหลัง หนังตาตก อาจมีหนังของนิ้วติดกัน |
| ถ่ายทอดทางกรรมพันธุ์แบบยีนด้อย (autosomal recessive) | ||
| กลุ่มอาการคาเพนเตอร์ (Carpenter) | ทุกรอยต่อ | ศีรษะรูปร่างผิดปกติ ร่วมกับมีจมูกแบน เอ็นหัวตาตก นิ้วเกิน |
2. กลุ่มที่มีรอยประสานกะโหลกศีรษะเชื่อมติดผิดปกติเพียงตำแหน่งเดียว (single-sutured craniosynostosis)
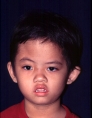
มักไม่มีความผิดปกติของส่วนอื่นในร่างกาย เนื่องจากไม่มีการกลายพันธุ์ของยีน จึงนิยมเรียกว่า non-syndromic craniosynostosis รอยประสานใดเชื่อมติดผิดปกติ ส่วนของกะโหลกศีรษะบริเวณนั้นก็จะขยายตัวไม่ได้ ศีรษะจึงมีรูปร่างบิดเบี้ยวเป็นแบบเฉพาะสำหรับความผิดปกติของแต่ละรอยประสาน เช่น ศีรษะยาวผิดปกติในแนวหน้าหลังคล้ายเรือ (scaphocephaly จาก sagittal craniosynostosis) ศีรษะรูปสามเหลี่ยม เป็นสันที่หน้าผาก (trigonocephaly จาก metopic craniosynostosis) ศีรษะเบี้ยว (plagiocephaly จาก unilateral coronal synostosis) สิ่งที่เราพบร่วมได้คือ ความผิดรูปของกะโหลกจะนำมาซึ่งการผิดรูปของใบหน้า หากไม่ได้รับการรักษาโดยเร็วในวัยทารก (รูปที่ 5)

การรักษา
ความผิดปกติแต่กำเนิดควรได้รับการดูแลทั้งทางกาย ใจ สังคม โดยสหวิชาชีพ ความผิดปกติทางร่างกายควรได้รับการแก้ไขโดยเร็วที่สุด โดยมีเงื่อนไขว่า การรักษาต้องไม่ทำให้เกิดผลเสียต่อผู้ป่วย หลักการในการรักษาภาวะกะโหลกศีรษะเชื่อมติดผิดปกติก็เช่นเดียวกัน ไม่แตกต่างจากโรคอื่น ๆ
กลุ่มที่มีรอยประสานกะโหลกศีรษะเชื่อมติดผิดปกติเพียงตำแหน่งเดียว
ปัจจุบัน มีบางโรงพยาบาลในประเทศที่มีอากาศหนาวเย็นทำการผ่าตัดตั้งแต่ผู้ป่วยอายุไม่กี่เดือน มีแผลผ่าตัดเล็กไม่กี่แผล ตัดกระดูกตรงรอยประสานที่มีการเชื่อมติดผิดปกตืทิ้งไปโดยมีกล้องส่องช่วย (endoscopic suturectomy) ตามด้วยการใส่หมวกที่ออกแบบมาเป็นพิเศษ (helmet) แต่ต้องใส่ทั้งวันเป็นเวลาหลายเดือน
ในประเทศไทยเราซึ่งมีอากาศร้อนชื้น การใส่หมวกดังกล่าวเป็นไปได้ยาก การผ่าตัดจึงยังเป็นวิธีมาตรฐานอยู่ คือ รอให้ผู้ป่วยมีอายุอย่างน้อย 6 เดือน จึงจะผ่าตัดปรับเปลี่ยนรูปร่างและขนาดกะโหลกศีรษะ จัดเป็นการผ่าตัดที่ใหญ่ เสียเลือดมากกว่า ต้องพักในไอซียูหลังผ่าตัด
ทั้งสองวิธี เราไม่ควรรอช้า ควรผ่าตัดแต่เนิ่น ๆ เพื่อหยุดการกดรัดเนื้อสมองใต้รอยประสานที่ผิดปกติ ถ้าเป็นศีรษะเบี้ยว (plagiocephaly) เนื่องจากรอยประสานโคโรนัลซึ่งอยู่เหนือหน้าผากเชื่อมติดผิดปกติเพียงข้างเดียว (unilateral coronal craniosynostosis) การผ่าตัดเร็วยังเป็นการป้องกันไม่มีเกิดใบหน้าบิดเบี้ยว (facial distortion) ซึ่งจะทำให้การแก้ไขยุ่งยากมากขึ้น อาจต้องมีการผ่าตัดซ้ำอีกเมื่อเด็กโตขึ้น
อย่างไรก็ตาม เราต้องระวังไม่ไปผ่าตัดให้ผู้ที่ไม่ได้เป็นโรคนี้ แต่มีศีรษะเบี้ยวจากแรงกดภายนอก (positional plagiocephaly หรือ deformational plagiocephaly) เช่น จากการที่ศีรษะถูกกดเบียดอยู่ในอุ้งเชิงกรานมารดาในขณะที่เป็นทารกในครรภ์
กลุ่มที่มีรอยประสานกะโหลกศีรษะเชื่อมติดผิดปกติหลายตำแหน่ง
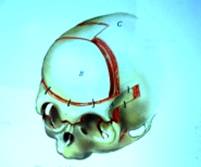
ในเรื่องการผ่าตัดกะโหลกศีรษะ วิธีส่องกล้องเพียงลำพังใช้ไม่ได้ในผู้ป่วยกลุ่มนี้ เพราะไม่เพียงพอที่จะเพิ่มพื้นที่ให้สมองได้ขยายตัว และไม่สามารถแก้ไขภาวะตาโปน ต้องผ่าตัดใหญ่ขยายกะโหลกศีรษะส่วนหน้าและเบ้าตา (frontoorbital advancement หรือ anterior cranial vault remodeling, รูปที่ 6) หรือขยายกะโหลกศีรษะส่วนหลัง (posterior cranial vault remodeling) หรือทั้งหน้าและหลัง
ต้องเข้าใจว่า การรักษาด้วยการผ่าตัดไม่ใช่ปลายทางของการดูแลผู้ป่วย หลังจากการผ่าตัด ยังต้องนัดผู้ป่วยมาดูเป็นระยะ ๆ เพื่อดูอาการ เช่น มีปัญหาแรงดันในสมองเพิ่มขึ้นหรือไม่ ตาโปนผิดปกติกลับขึ้นมาอีกหรือไม่ มีปัญหาการหายใจอันเนื่องมาจากใบหน้าส่วนกลางไม่เจริญเติบโตทำให้ทางเดินหายใจแคบหรือไม่ (รูปที่ 7) มีปัญหาเรื่องการเคี้ยวอาหารอันเนื่องมาจากการสบฟันที่ผิดปกติหรือไม่ เพื่อจะได้ดำเนินการแก้ปัญหาต่อไป


